

Instructions
Welcome to CTREP-finder 1.0
To feed 10 billion people in 30 years from now will heavily depend on new higher yielding, yet safe, biotech crop varieties. Biotech varieties are crops developed using biotechnologies, particularly genetic transformation and genome editing. A clean transgenic plant is a plant that contains only the defined foreign DNA fragment (transgene) in a defined genomic location, while a clean gene-edited plant is a plant free of any foreign DNA. Here we present the CTREP-finder, a user-friendly web server for fast detection, characterization and visualization of any foreign DNA fragment in TREPs, based on next-generation whole genome sequencing data. This server facilitates the rapid selection of clean transgenic or edited plants (CTREPs) for crop breeding programs, thus clear the biosafety concerns relating to plasmid backbone fragment insertions, with little requirement for bioinformatics expertise.
Analysis
How to complete the identification of CTREPs?
The Analysis includes the identification and localization of transgenes. First, the system determines whether (i) an edited plant is free of transgenes and (ii) transgenic plants are free of backbone DNA. Second, the system helps determine the location of the transgene(s) in plant genome.
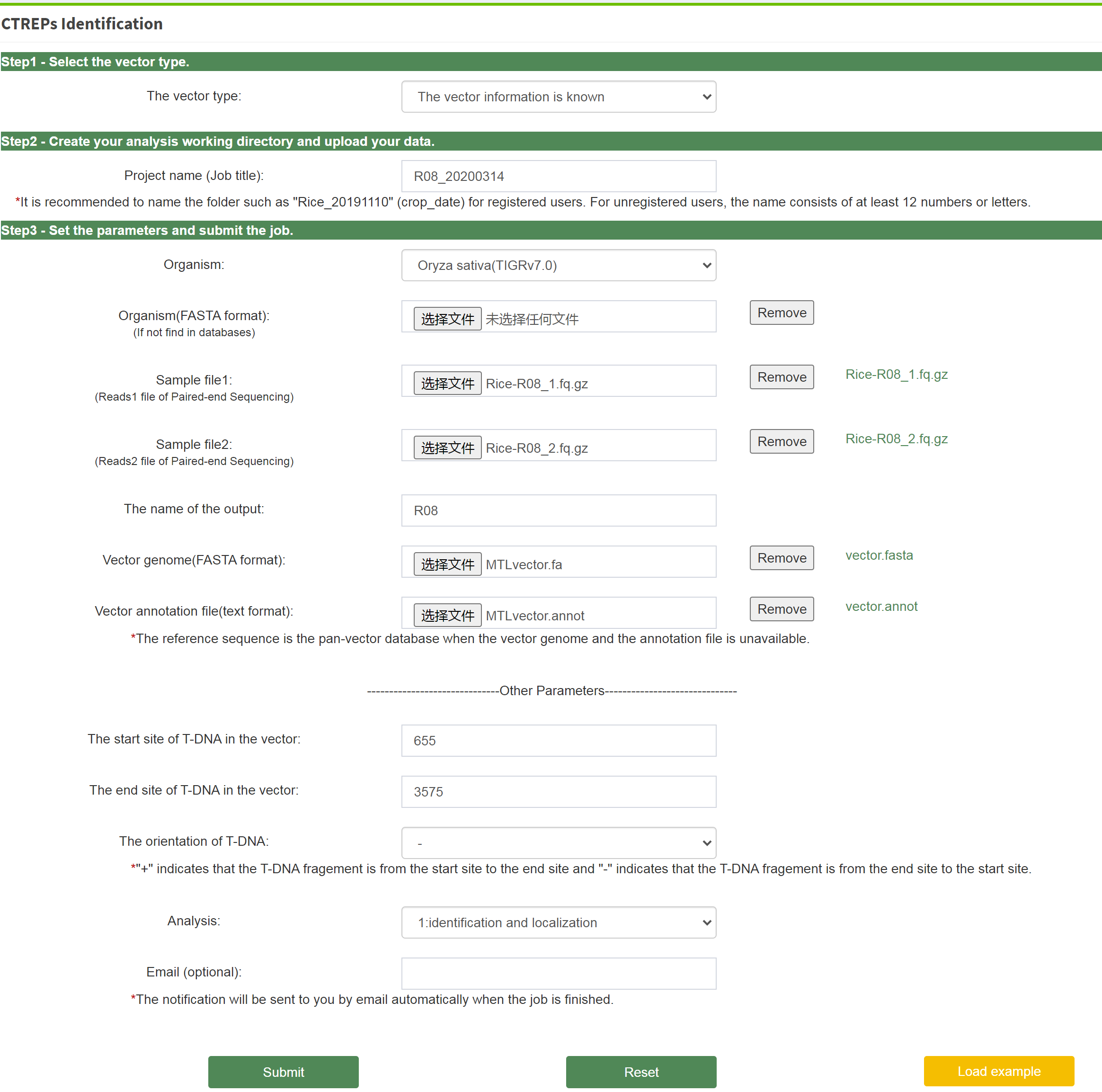
Once logged into the analysis platform of CTREP-finder as default, Users need to select the vector type in the first step. The required parameters and analyis pipelines used for identification of known or unknown vectors are quite different. Users are allowed to create a project name (job title). It is recommended to name the folder such as "Rice_20191110" (crop_date) for registered users. For unregistered users, the name that consists of at least 12 numbers or letters to avoid data confusion is appreciated. CTREPs identification with known vector, the raw sequencing files, plant genome, the vector genome, the annotation file of vector, the start site and end site of T-DNA on the vector is required, as well as the orientation of the T-DNA strand. The direction of this T-DNA strand is indicated by “+” or “-”, where + indicates that the the interval of T-DNA is from start site to the end site; - indicates that the T-DNA's interval is from the end site to the start site. It also supports users to upload their own plant genome when it is not found in our plant genome database. Prepared all of above files, users need to select the analysis content and click the "submit" button to start the analysis job at last.
The program is executed and the result report is generated automatically, once a job is submitted successfully. Users can visit the following website to view and download the results later. The notification and results will be sent to users by email automatically when the job is finished and the email is told.
It is advised to login with registered account, if users want to save the inputs and outputs, or check the mapping information, or manage the analysis project.
What format of input files is required?
The sequencing files is the paired-end sequencing compressed files in the fastq format from the next-generation sequencer, the vector reference sequences and the plant genome sequences should be in the FASTA format (the suffix of file name is .fa or .fasta). The first line starts with a symbol ">", followed by sequence name and description. The second line is the sequence, containing only alphabets for each nucleoside.
The annotation file with TXT format contains four columns: The first column is the chromosome of the vector. Since the vector has only one sequence, the default is chr1 here. The second and third columns are the start site and end site of genes on the vector, respectively, and the fourth column is the gene name. Less detailed gene annotation of the vector, for example, which only includes the position of LB and RB, is also allowed. However, more details are favored and will lead to more information in results.
What should I do if I don't have the vector annotation file?
Users can copy the vector information in FASTA format, paste them into the blank column of vector identification module, and click the "Submit" button, the annotation figure and annotation TXT file in blast m8 format will be automatically generated for user reference. Finally, it's simple and easy to convert the blast m8 format file into the required format of vector annotation.
Is there any examples for analysis?
Yes, users can download the exmaple data from the webpage, load them in the appropriate place, select the vector type, fill in the project name and Email (optional). After that, click the button of "Load example" and "Submit" to start. The results and report will be generated several minutes later.
What are the criteria used for determining a plant as CEP or CTP?
A clean edited plant (CEP) is a plant with neither T-DNA nor any backbone fragment detected in the sequencing data. The criteria used are:
(i) VRN≥Gdepth/2+VectorLen/ReadLen, and Tcov≥0.9, for plants with known plasmid;
(ii) FRN≥ (Gdepth/2)×10, combined with characteristic structure ie., CaMV35S or OsCas9 was identified, for plants without any background information.
VRN is the number of reads that matched the vector. Gdepth, Tdepth and Bdepth represent the average sequencing depth of the recipient plant genome, T-DNA, and the backbone; VectorLen is the sequence length of the vector; ReadLen is the length of each read; Tcov is the base coverage of the T-DNA; FRN is the number of reads that match the pan-vector databases.If a vector consists of host plant genome sequences, these sequences should be excluded.
Which plants are included in the plant genome database?
At present, 24 plant species are suported in the plant genome database which were downloaded from Phytozome (https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Phytozome). They were listed as following:
| Species | Version | Species | Version | Species | Version | Species | Version |
|---|---|---|---|---|---|---|---|
| Arabidopsis thaliana | TAIR10 | Brassica napus | v2.0 | Brassica oleracea L. | v1.0 | Brassica rapa | v1.3 |
| Carica papaya | ASGPBv0.4 | Citrus sinensis | v1 | Cucumis sativus L. | v1.0 | Glycine max | v2.0 |
| Gossypium hirsutum | v2.0 | Gossypium raimondii | v2.0 | Hordeum vulgare | v1.0 | Malus domestica | v1.1 |
| Oryza sativa | v7.0 | Pyunus persica | v2.0 | Ricinus communis | TIGRv0.1 | Selaginella moellendorffii Hieron. | v1.0 |
| Setaria italica | v2.0 | Sorghum bicolor | v3.0 | Solanum lycopersicum | v3.0 | Solanum tuberosum L. | v4.03 |
| Triticum aestivum L. | v2.0 | Vigna unguiculata | v1.0 | Vitis vinifera L. | v2.0 | Zmays_284 | 284AGPv3,PH207v1.0 |
JBrowse visualization
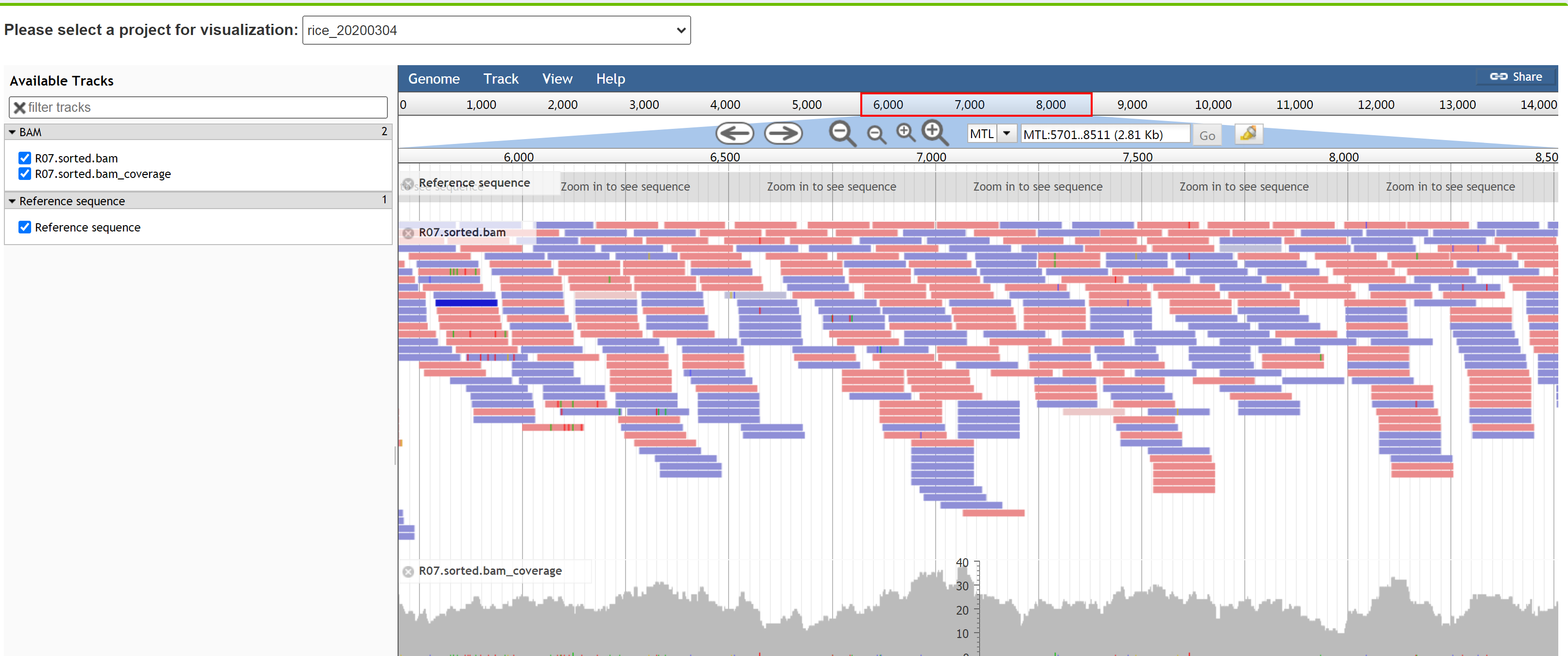
We provided the JBrowse visualization for users' convenience to retrieve the mapping information of reads with the expression vector sequence. JBowse is a genome browser which is developed based on HTML5 and Javascript language. Fast, smooth scrolling and zooming is supported. The small rectangular with colors represents one read. The colors represent the mapping status of the paired-end reads. Light red means both of the paired-end reads were mapped to the reference sequence, and this is the left read. The right read is colored with light blue. Dark read means the left read that was mapped to the reference sequence with the right read was unmapped. Dark blue means the right read that was mapped to the reference sequence with the left read was unmapped. The letters in the small rectangular are considered as a real SNP, as well as a random error from sequencer.
Vector Databases
How the pan-vector database was constructed?
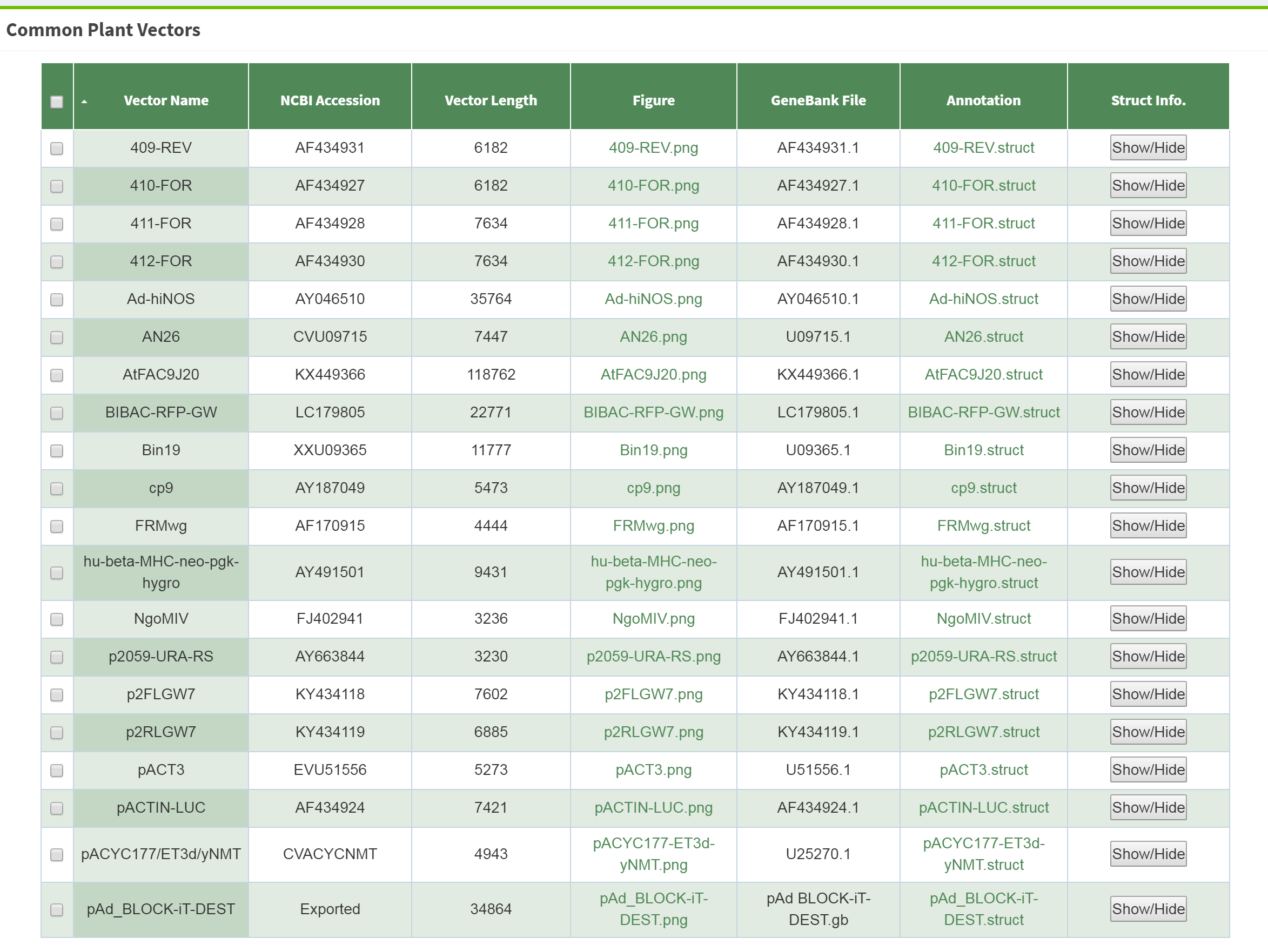
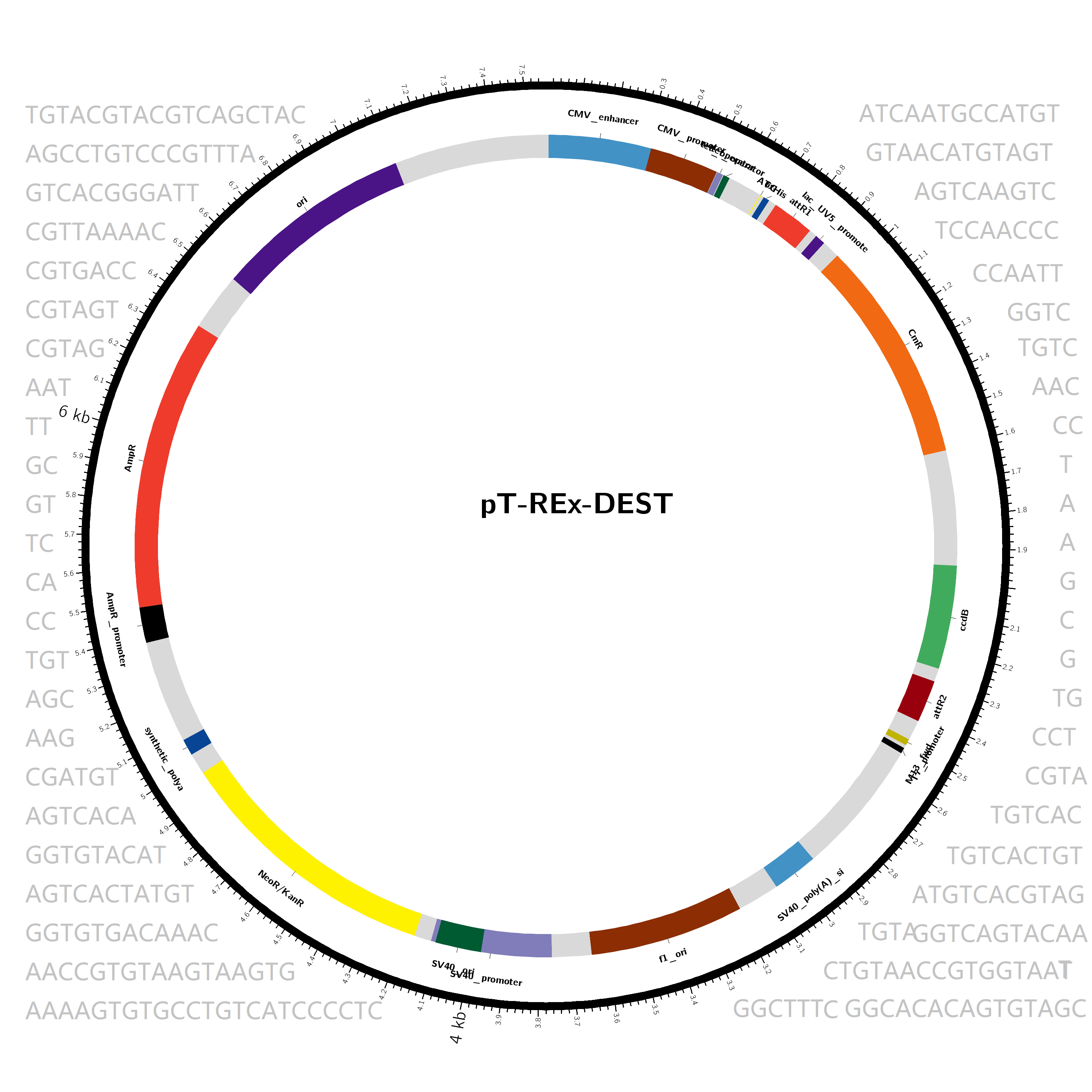
The raw data component of the pan-vector database was constructed by searching the keywords "gateway vector/plasmid", "plant vector/plasmid" on NCBI (https://www.ncbi.nlm.nih.gov/), downloading the results in genebank format, then combining thesewith the plant vectors part of addgene (https://www.addgene.org/) and other common plant vectors from the literature. We performed the following steps with in-house scripts before the tens of thousands of individual data items were imported into the database. First, duplicate vectors, incomplete vectors and vectors less than 1kb in length were filtered through this process. Second, the gene annotation of the vectors was unified, when inconsistent description on the same gene was found, and the feature index table was built according to the annotation. The diagram for each vector was then drawn and the feature sequences that have no homology with the plant genome under study were stored in FASTA format. Finally, a total of 1095 genes and 1388 vectors are kept.
What algorithm was used for CTREPs identification with unknown vector?
When transgenic sequences are identified in a sample using the pan-vector database, a candidate vector is predicted based upon characteristic structures detected in the raw sequencing data of the sample. The CTREP-finder searches for these characteristic structures in the pan-vector database to generate a report including the top ten predictions (hits), each with a score and an expectation value defining its probability and confidence. The smaller the expected value, the more likely the unknown sequence will be considered the predicted vector.
Does it support users to search and upload plant vector information that isn't included in the pan-vector database?
Yes. We also designed the search and upload module in the program to automatically search and compare the submitted vector sequences with the existing vectors in the pan-vector database. When a new vector is spotted, a review process will ensue and the valid vector sequence will be added to the database. Uploading and sharing the new vector is appreciated.
About results
How to understand the analysis results?
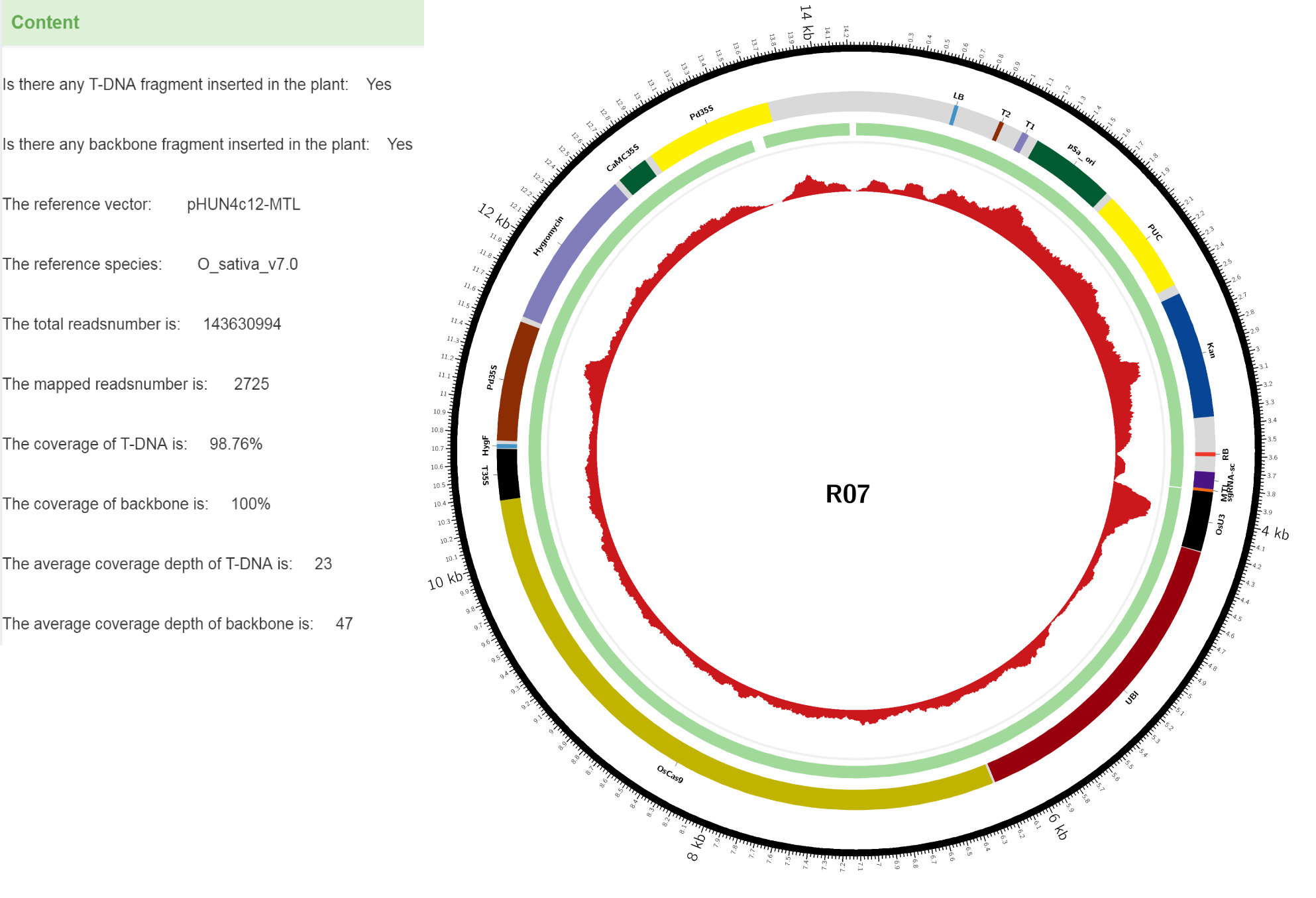
Take a gene edited plant as an example. R07 is a gene-edited rice (Oryza sativa L.) plant. It was developed by transforming the CRISPR/Cas9 vector pHUN4c12S-MTL (14251 bp) into rice cultivar Xidao No.1 via Agrobacterium-mediated transformation. pHUN4c12S-MTL was modified from pHUN4c12S by replacing the sgRNA sequence targeting the third exon of LOC_Os03g27610 (OsMTL), the sole homolog of maize MTLgene. The resulting T1 mutant plants were identified by sequencing the target fragment of OsMTL. The genome of one such T1 mutant plant was sequenced by Illumina NovaSeq6000 based on the WGS protocol.
Figure 1 shows the vector (pHUN4c12S-MTL) used for transformation and the reads identified in the plant.The outer layer shows the length scale of the vector. The second layer is the annotation of genes and components of the vector. The third layer represents the coverage interval of the expression vector detected in sequencing. The fourth layer is the depth of each base of the vector detected in sequencing.The results show that both the T-DNA and backbone are present in the edited plant.
Localization analysis shows that the T-DNA fragment and backbone DNA are indeed integrated into two different chromosomes, i.e. the backbone DNA is on chromosome 1 and the T-DNA is on chromosome 10 (Figure 2). Detailed information is given in a txt file (Table 1), which shows the exact site of respective insertion, and that deletions occurred in both the left border (LB, a 6-bp deletion) and right border (RB, completely deleted). Furthermore, the backbone sequence, including the RB border, was homozygous and had been integrated into Chromosome 1 of the rice genome according to the average depth of T-DNA and backbone. The upstream and downstream 500bp sequences flanking the integration sites are also provided in the txt file to facilitate users' PCR verification. The integrated sequences and sites can also be checked in the JBrowse.
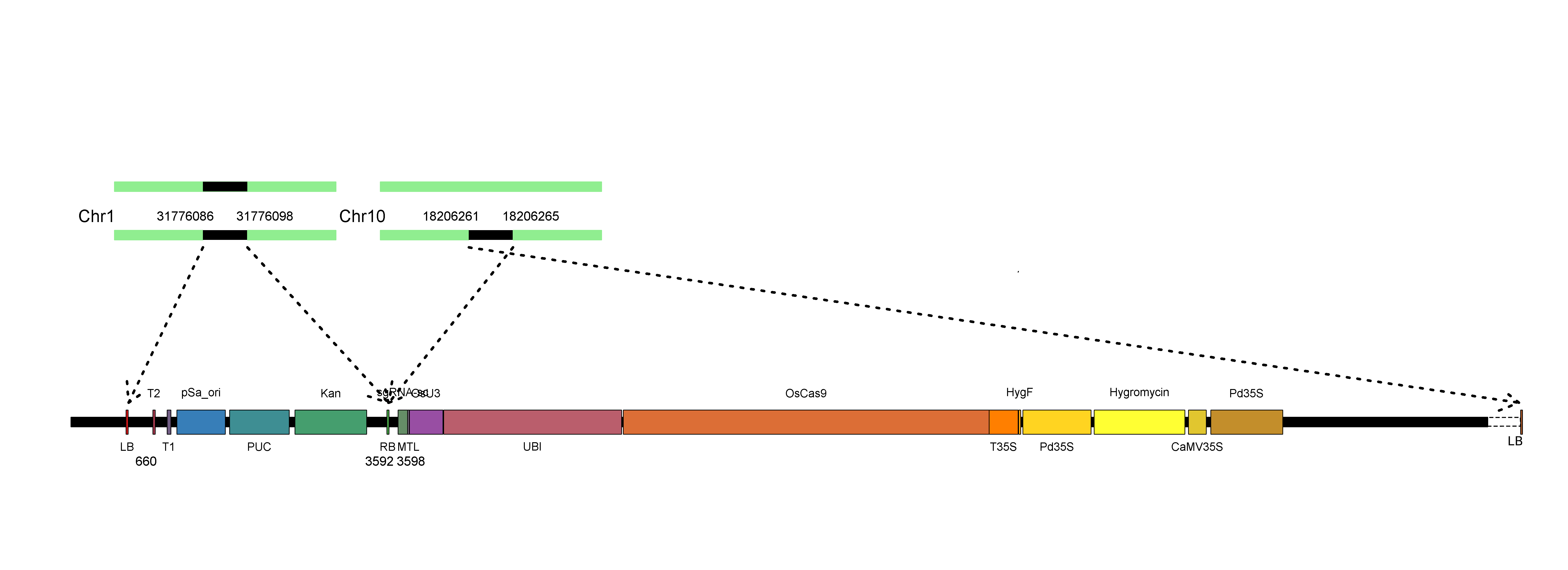
| Sample | VectorName | VectorLength | VecotrStrand | VectorBreakpoint | Genome | Chromosome | ChromLength | GenomeBreakpoint | Genomeend | GenomeStrand | LeftSeq | RightSeq |
| R07 | MTLvector | 14251 | - | 3592 | O_sativa.v7 | Chr1 | 43270923 | 31776098 | 31776598 | + | - | The downstream 500bp of genomic integrated site |
| R07 | MTLvector | 14251 | + | 660 | O_sativa.v7 | Chr1 | 43270923 | 31776086 | 31775586 | - | The upstream 500bp of genomic integrated site | - |
| R07 | MTLvector | 14251 | - | 649 | O_sativa.v7 | Chr10 | 23207287 | 18206261 | 18205761 | - | The downstream 500bp of genomic integrated site | - |
| R07 | MTLvector | 14251 | + | 3598 | O_sativa.v7 | Chr10 | 23207287 | 18206265 | 18206765 | + | - | The downstream 500bp of genomic integrated site |
Data Management
Users can access to the data management window after registration/login, which serves two functions: management and projects. For storage,each user is provided with a 300-GB space. Availability and used spaces are represented by green and red colors, respectively. When available space is less than 10%, a cleanup on existing data is required to make enough space for data analysis and processing, otherwise the CTREP-finder cannot work. Click the "Delete" button at the end of the selected folder to remove the data. The cleanup is irreversible. Please be advised to download and save the data to avoid any loss. For project management, the status of individual task is shown, and once the project completed, the results are available for download under the project lists.
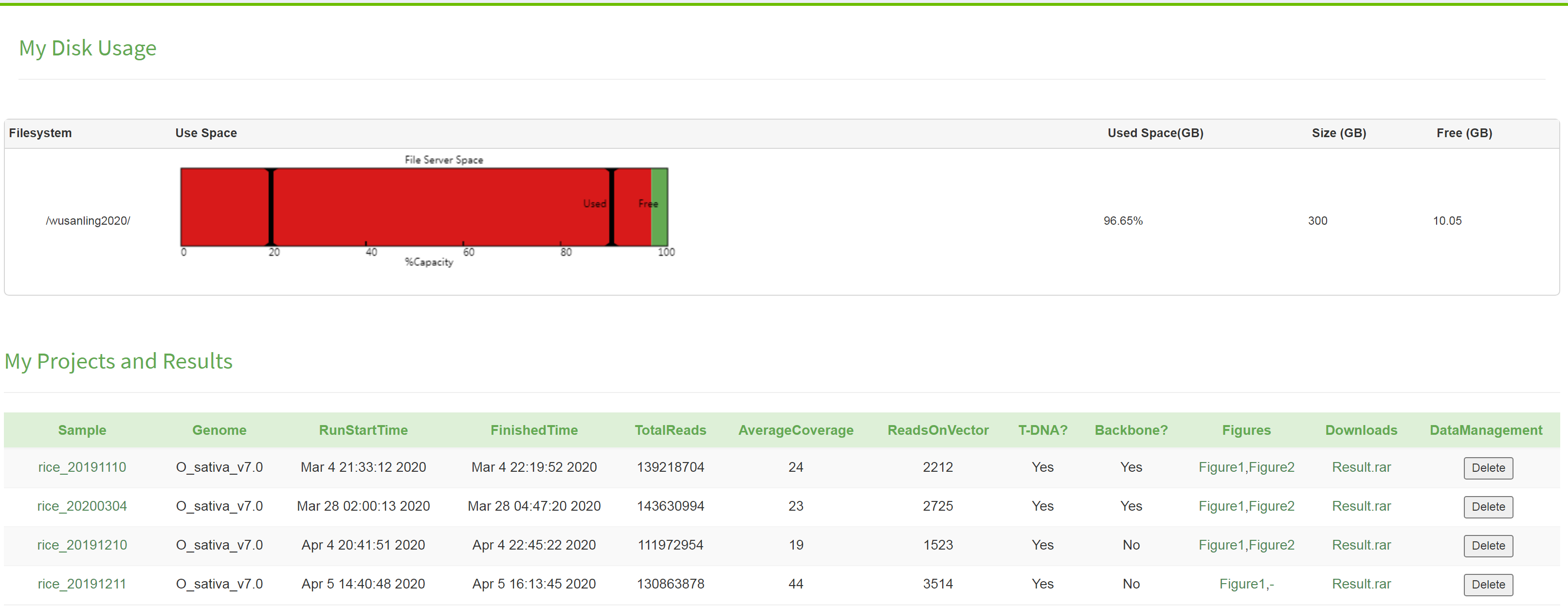
How to manage my storage spaces and review projects?
As shown above, the user has already used 290GB from the 300-GB data space. The user performed four projects: with the results shown, which can be downloaded. For the rice project, identification of trangenic/gene editing has completed and the results are available. The Figure 1 in the graphic report is the identification diagram of trangenic/gene editing. However, if the Figure 1 is represented by the symbol "-", the trangenic/gene editing identification is either unscheduled or still ongoing. The Figure 2 is the insertion location diagram of trangenic/gene editing. Likewise, if the Figure 2 is represented by the symbol "-", the trangenic/gene editing insertion location analysis is either unscheduled or still ongoing.
How long does CTREP-finder take to perform the identification and localization analysis of CTREPs?
For Oryza sativa with a 374MB reference genome size, 50×seuencing coverage depth and 40GB raw data, the results can be obtained within 2 hours. It takes a little more time for plants with large genome such as barley etc.
How to open files with the suffix of .out in Result.rar?
The files with the suffix of .out can be opened through Notepad or Microsoft Office Excel.
How about browser compatibility?
The service has been successfully tested in Google Chrome, Mozilla Firefox, Safari, Microsoft Edge, on Linux, MacOS, and Windows operating system.
What should I do if I have trouble in use?
Please contact the administrator: biomics@zju.edu.cn
Do you have a video tutorial on how the website works?
Yes, the link for the tutorial is here:CTREP-finder tutorial
References
1. Gaj, T., Gersbach, C.A. and Barbas, C.F. (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol, 31, 397-405.
2. Urnov, F.D., Rebar, E.J., Holmes, M.C., Zhang, H.S. and Gregory, P.D. (2010) Genome editing with engineered zinc finger nucleases. Nat Rev Genet, 11, 636-646.
3. Bhatnagar-Mathur, P., Vadez, V. and Sharma, K.K. (2008) Transgenic approaches for abiotic stress tolerance in plants: retrospect and prospects. Plant Cell Rep, 27, 411-424.
4. Zhang, Y., Massel, K., Godwin, I.D. and Gao, C.X. (2018) Applications and potential of genome editing in crop improvement. Genome Biol, 19.
5. Hua, K., Zhang, J.S., Botella, J.R., Ma, C.L., Kong, F.J., Liu, B.H. and Zhu, J.K. (2019) Perspectives on the Application of Genome-Editing Technologies in Crop Breeding. Mol Plant, 12, 1047-1059.
6. Kononov, M.E., Bassuner, B. and Gelvin, S.B. (1997) Integration of T-DNA binary vector 'backbone' sequences into the tobacco genome: Evidence for multiple complex patterns of integration. Plant J, 11, 945-957.
7. Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Proc, G.P.D. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078-2079.
8. Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman, D.J. (1990) Basic Local Alignment Search Tool. J Mol Biol, 215
9. Bolger, A.M., Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114-2120.
10. Huang, X.Q. and Madan, A. (1999) CAP3: A DNA sequence assembly program. Genome Res, 9, 868-877.
11. Skinner, M.E., Uzilov, A.V., Stein, L.D., Mungall, C.J. and Holmes, I.H. (2009) JBrowse: A next-generation genome browser. Genome Res, 19, 1630-1638.
12. Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., Jones, S.J. and Marra, M.A. (2009) Circos: An information aesthetic for comparative genomics. Genome Res, 19, 1639-1645.